2 min to read
基于GATK检测基因组SNP和indel
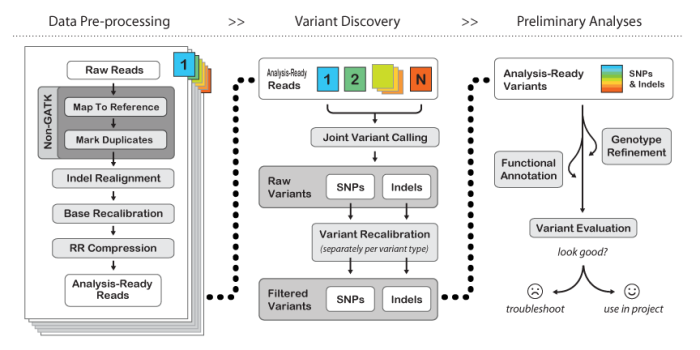
- content {:toc}
概念
二代测序原理
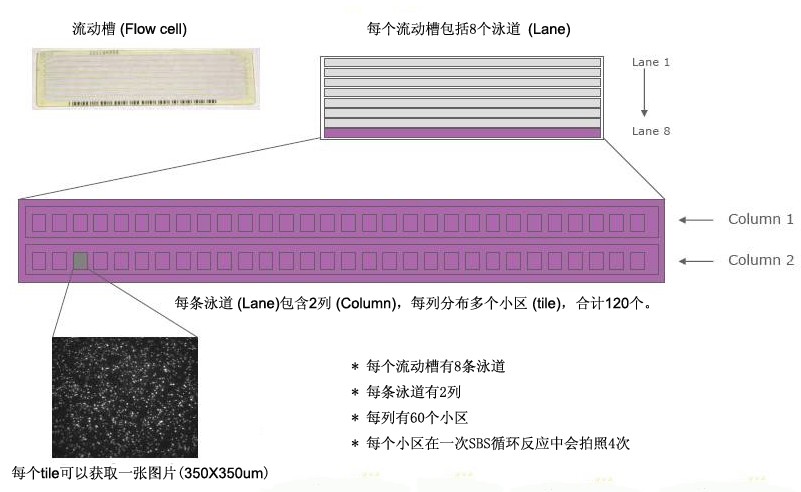

Illumia测序原理简单概括就是将文库结合到测序芯片上,并通过PCR将单一序列扩增成簇以提高信号强度,然后测序时收集每一簇的荧光信号,并转换为相应的碱基,从而获取测序数据。
SBS:边合成边测序反应
Run:单次上机测序反应
Lane:单泳道,每条泳道可以直接物理区分测序样品
Tile:小区,每条Lane中排有2列tile,合计120个小区。每个小区上分布数目繁多的簇结合位点。
Cluster:簇,在Solexa测序技术中会采用桥式PCR方式生产DNA簇,每个DNA簇才能产生亮度达到CCD可以分辨的荧光点。
Index:标签,在Solexa多重测序(Multiplexed Sequencing)过程中会使用Index来区分样品,并在常规测序完成后,针对Index部分额外进行7个循环的测序,通过Index的识别,可以在1条Lane中区分12种不同的样品。
hard filter和VQSR
hard filter和VQSR为原始变异检测过滤的两种不同方法,前者是通过GATK的VariantFiltration完成,后者是通过GATK的VQSR(变异位点质量值重新校正)进行过滤。
VQSR是根据已有的真实变异位点(人类基因组一般使用HapMap3中的位点,以及这些位点在Omni 2.5M SNP芯片中出现的多态位点)来训练,最后得到一个训练好的能够很好的评估真伪的错误评估模型。使用VQSR数据量一定要达到要求,数据量太小无法使用高斯模型。一个位点是真实的概率比上这个位点可能是假阳性的概率的log odds ratio(对数差异比),因此,可以定性的认为,这个值越大就越好。
所需工具
- sratoolkit
- bgzip
- tabix
- bwa
- samtools
- GATK
分析流程
数据下载
下载E.coli K12的参考基因组序列
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/005/845/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna.gz
gzip -dc GCF_000005845.2_ASM584v2_genomic.fna.gz > E.coli_K12_MG1655.fa
samtools faidx E.coli_K12_MG1655.fa #创建索引
下载E.coli K12的测序数据
fastq-dump --split-files SRR1770413.sra
比对
#为参考序列构建BWA比对所需的FM-index
bwa index E.coli_K12_MG1655.fa
#比对
bwa mem -t 4 -R '@RG\tID:foo\tPL:illumina\tSM:E.coli_K12' E.coli_K12_MG1655.fa SRR1770413_1.fastq.gz SRR1770413_2.fastq.gz | samtools view -Sb - > E_coli_K12.bam && echo "** bwa mapping done **" #@RG含义参考[5]
#排序
time samtools sort -@ 4 -O bam -o E_coli_K12.sorted.bam E_coli_K12.bam && echo "** BAM sort done"
#删除不必要文件
rm -f E_coli_K12.bam
#3 标记PCR重复
time ~/software/gatk-4.0.11.0/gatk MarkDuplicates -I E_coli_K12.sorted.bam -O E_coli_K12.sorted.markdup.bam -M E_coli_K12.sorted.markdup_metrics.txt && echo "** markdup done **" #去除这些由PCR扩增所形成的duplicates
#4 删除不必要文件
rm -f /Project/201802_wgs_practice/output/E.coli/E_coli_K12.bam
rm -f /Project/201802_wgs_practice/output/E.coli/E_coli_K12.sorted.bam
#5 创建比对索引文件
time samtools index E_coli_K12.sorted.markdup.bam && echo "** index done **"
变异检测
~/software/gatk-4.0.11.0/gatk CreateSequenceDictionary -R E.coli_K12_MG1655.fa -O E.coli_K12_MG1655.dict && echo "** dict done **"
#1 生成中间文件gvcf
time ~/software/gatk-4.0.11.0/gatk HaplotypeCaller \ #能过通过对活跃区域(也就是与参考基因组不同处较多的区域)局部重组装,同时寻找SNP和INDEL。[7]
-R E.coli_K12_MG1655.fa \
--emit-ref-confidence GVCF \
-I E_coli_K12.sorted.markdup.bam \
-O E_coli_K12.g.vcf && echo "** gvcf done **"
#2 通过gvcf检测变异
time ~/software/gatk-4.0.11.0/gatk GenotypeGVCFs \
-R E.coli_K12_MG1655.fa \
-V E_coli_K12.g.vcf \
-O E_coli_K12.vcf && echo "** vcf done **"
bgzip -f E_coli_K12.vcf #压缩
tabix -p vcf E_coli_K12.vcf.gz #构建tabix索引
# 使用SelectVariants,选出SNP
time ~/software/gatk-4.0.11.0/gatk SelectVariants \
-select-type SNP \
-V E_coli_K12.vcf.gz \
-O E_coli_K12.snp.vcf.gz
# 为SNP作硬过滤
time ~/software/gatk-4.0.11.0/gatk VariantFiltration \
-V E_coli_K12.snp.vcf.gz \
--filter-expression "QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \
--filter-name "Filter" \
-O E_coli_K12.snp.filter.vcf.gz
# 使用SelectVariants,选出Indel
time ~/software/gatk-4.0.11.0/gatk SelectVariants \
-select-type INDEL \
-V E_coli_K12.vcf.gz \
-O E_coli_K12.indel.vcf.gz
# 为Indel作过滤
time ~/software/gatk-4.0.11.0/gatk VariantFiltration \
-V E_coli_K12.indel.vcf.gz \
--filter-expression "QD < 2.0 || FS > 200.0 || SOR > 10.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \
--filter-name "Filter" \
-O E_coli_K12.indel.filter.vcf.gz
# 重新合并过滤后的SNP和Indel
time ~/software/gatk-4.0.11.0/gatk MergeVcfs \
-I E_coli_K12.snp.filter.vcf.gz \
-I E_coli_K12.indel.filter.vcf.gz \
-O E_coli_K12.filter.vcf.gz
参考资料
[3]. 从零开始完整学习全基因组测序(WGS)数据分析:第3节 数据质控
[4]. alignment-and-variant-calling-tutorial
[5]. GATK sample/library/lane meaning in BAM read group @RG
[6]. GATK使用方法详解(原始数据的处理)

Comments